


Schizophrenia is a complex disorder (or, more likely, group of disorders) that has reality distortion at its core. Efforts to establish the cause of schizophrenia have been ongoing for more than a century, and many models have come and gone in that time (not for nothing has schizophrenia been called ‘the graveyard of neuropathologists’ (Plum, 1972)).
Part of the problem has been that very often the models would deal adequately with one aspect of the disorder (the neurochemistry, say, or the cognitive impairment) but fail to explain another, or just ignore it. Without a good model of schizophrenia, developing ways to treat the condition or prevent it before it begins is much harder, and much less likely to be successful.
In an article published in The Lancet at the end of last year, Oliver Howes and Robin Murray have set out what they call ‘an integrated sociodevelopmental-cognitive model’. As can no doubt be told from that title, this is a model that tries to account for the very wide range of factors that have been associated with increased risk for psychosis, from the neurobiological to the socio-demographic.

At root, this model suggests that the key biological dysfunction in people with schizophrenia is that they make and release too much of the neurotransmitter dopamine. This is not new (dopamine models of schizophrenia have been around since the first antipsychotics were shown to block dopamine receptors) but Howes and Murray tie this together with another well-established model, that of schizophrenia being a neurodevelopmental disorder.
In the original neurodevelopmental model, proposed by Murray (Murray and Lewis, 1987) and others (Weinberger, 1987) nearly 30 years ago, altered dopamine was a result of the interaction between early damage to frontal brain regions (perhaps as the result of complications during birth) and normal development.
Now, Howes and Murray argue events early in life, such as birth complications but also now including the effect of genes on brain development and childhood adversity, sensitise the dopamine system. This sensitisation makes the system more vulnerable to stress, such as that caused by social adversity. At the same time, cognitive models suggest that such stress can bias an individual’s thinking toward paranoia. A vicious circle develops, where the symptoms result in greater social adversity and stress, leading to more symptoms and so on, the end result being the full threshold psychotic break.
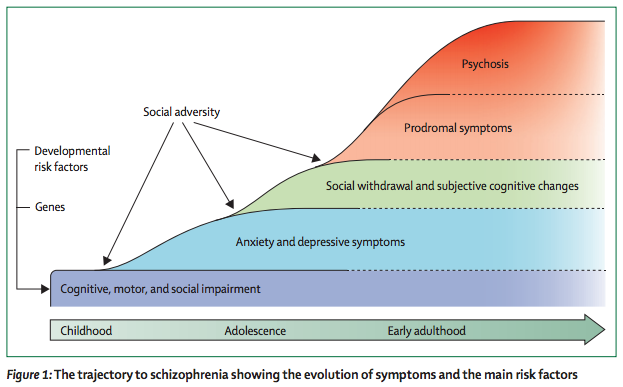
This model represents a real advance, in that it puts life events and the impact they have on the individual’s interpretation of the world around them at the centre of the process leading to schizophrenia. This fits with much of the advances of the past decade or so, that show that being an immigrant or being exposed to physical or sexual abuse are associated with a threefold increase in risk. It also suggests that interventions to reduce stress, or alter disordered patterns of thinking (such as cognitive behavioural therapy), ought to be particularly beneficial in the early phases of illness.
However, the article is not without problems. Largely, this is because the authors are not trying to conduct a systematic review, but to bolster the case for their model. In several places, the evidence is rather weaker than is made out, or is heavily reliant on animal models that may not in the end prove applicable to the human. Howes and Murray themselves concede that their model does not easily account for the persistent negative symptoms and cognitive impairments seen in many patients.
For me though, the major concern is whether a single model can ever fit a heterogenous disorder such as schizophrenia, or whether it is more likely to explain some fraction of the population with the diagnosis.
One example will serve to explain this point – Howes and Murray focus almost exclusively on dopamine as the neurotransmitter responsible for psychotic symptoms. However, we already know that many patients do not respond to treatment with traditional, dopamine-targeting, antipsychotics (Pilowsky, 1993). Indeed, there is evidence that non-responding patients show alterations in another neurotransmitter, glutamate, instead (Egerton, 2012). Still other reports indicate a subset of patients with changes to a specific group of acetylcholine receptors (Gibbons, 2013).
Better characterising the subgroups of patients with schizophrenia is a critical condition of establishing the success or failure of this new model.

Links
Howes OD and Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. The Lancet – 6 December 2013. DOI: 10.1016/S0140-6736(13)62036-X. [Abstract]
Plum F. Prospects for research on schizophrenia. Neuropathological findings. Neurosci Res Prog Bull. 1972;10:384–388. [PubMed abstract]
Murray RM and Lewis. Is schizophrenia a neurodevelopmental disorder? British Journal of Psychiatry 1987;295:681-682.
Weinberger DR. Implications of Normal Brain Development for the Pathogenesis of Schizophrenia. Archives of General Psychiatry 1987;44:660-669. [PubMed abstract]
Pilowsky LS et al. Antipsychotic medication, D2 dopamine receptor blockade and clinical response: a 123I IBZM SPET (single photon emission tomography) study. Psychological Medicine 1993;23:791-797. [PubMed abstract]
Egerton et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology 2012;37:2515-2521.
Gibbons et al. Widespread decreases in cortical muscarinic receptors in a subset of people with schizophrenia. 2013;16:37-46. [PubMed abstract]




https://www.nationalelfservice.net/mental-health/schizophrenia/a-new-model-for-schizophrenia/ – Site Title
8 years agoAnne
9 years agoAnd_Cipriani
12 years agoMedLinkNeurol
12 years agoPusiGalor
12 years agoPsyPositive
12 years agoigorthiriez
12 years agoLydia Watson
12 years agoHisKingdom4Ever
12 years agopsynthesisblog
12 years agocsmony
12 years agoNatashaSloman
12 years agokrysiacanvin
12 years agoFutureDrWolfe
12 years agodenisthehat88
12 years agoMental_Elf
12 years agosoniajohnson
12 years agoVivien Kemp
12 years agoAnneka Holden
12 years agoJoão Leal
12 years agoClinpsychLucy
12 years agoVMorfee
12 years agopatmmc2
12 years agoSameiHuda
12 years ago444blackcat
12 years agoForensicDetail
12 years agoLightbulbPsych
12 years agoedoyap
12 years agoShonaH1961
12 years agomarinkestassen
12 years agoJimmyscambridge
12 years agoDLapthorne
12 years agomonarisa83
12 years agoMental_Elf
12 years agodaceituno
12 years agoLisamarsh100
12 years agoDiasporicLife
12 years agoDr_Asrat
12 years ago121Therapy
12 years ago121Therapy
12 years ago121Therapy
12 years agoLizHughesDD
12 years agoMental_Elf
12 years agoFrankCurtisLib
12 years agoBernadette1708
12 years agoVansVoice
12 years agoHaHaStayinAlive
12 years agoClinPsychology
12 years agoTheChildElf
12 years agoJen_ICCR
12 years agoMental_Elf
12 years agoBespoke Training Services UK Ltd
12 years agoThe Mental Elf
12 years agoHHLibService
12 years agoPLUSBarnet
12 years agoaghoury79
12 years agoSCEW86
12 years agoBTSUKLTD
12 years agobsspsychology
12 years agolypftlib
12 years agomolloy_p
12 years agoMental_Elf
12 years agolyhairian
12 years agoScott Inglis
12 years agoBwoodHighland
12 years agoPagan Warren
12 years agoPaula Gardiner
12 years agoMarjorie Camus Charron
12 years agoSunrise_Glen
12 years agoPagan Warren
12 years agojulie_hankin
12 years agoKevinMacGlashan
12 years agoCharlotte Rowe
12 years agoaghoury79
12 years agoDavid_K_says
12 years agoCShawAiW
12 years agosamirshah
12 years agoCShawAiW
12 years ago121Therapy
12 years agoian_hamilton_
12 years ago